
EPJ B Colloquium - Next generation interatomic potentials for condensed matter systems
- Details
- Published on 08 July 2014
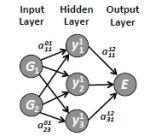
More efficient computational methods are urgently needed to capture condensed matter systems in simulations. Electronic structure methods, such as density-functional theory (DFT), usually provide a good compromise between accuracy and efficiency, but they demand much computational power. For this reason, they are only applicable to small systems containing a few hundred atoms at most. Conversely, many interesting phenomena involve much larger systems comprising thousands of atoms or more. Considerable effort has been invested in the development of potentials that enable simulations to run on larger system and for longer times. Typically these potentials are based on physically-motivated functional forms. Therefore, while they perform very well for the specific applications for which they have been designed, they cannot easily be transferred from one system to another. Moreover, their numerical accuracy is restricted by the intrinsic limitations of the imposed functional forms. In this EPJ B Colloquium, Handley and Behler survey several novel types of potentials emerged in recent years, which are not based on physical considerations. Instead, they aim to reproduce a set of reference electronic structure data as accurately as possible by using very general and flexible functional forms. While they differ in the choice of the employed mathematical functions, they all provide high-quality potential energy surfaces, and their efficiency is comparable to conventional empirical potentials. In many cases these potentials now offer a very interesting new approach whereby complex systems may be studied with unprecedented accuracy.
Christopher M. Handley and Jörg Behler (2014), Next generation interatomic potentials for condensed systems, European Physical Journal B, 87: 152, DOI: 10.1140/epjb/e2014-50070-0



